Contributing to the development of scientific software provides a viewpoint on the process by which new algorithms are conceived, and translated in efficient and usable libraries, a deep understanding of an area of computational materials modeling, and practical skills in solving research with problems with code. For the Spring Semester 2024 we propose to contribute to implement new features, and/or demonstrate their use in materials modeling problems, for one of the packages that are actively developed on our github page. Below you can find some examples. We invite you however to look directly at the github pages and the open issues, and if you find something that interests you we may be able to turn that into a semester project. The details and the scope of the research plan will be discussed with the student, and adapted depending on the level of prior knowledge and whether it is at the Master or Bachelor level.
Automatic Recognition of Molecular Motifs
Most of the current understanding of structure−property relations at the molecular and the supramolecular scales can be formulated in terms of the stability of and the interactions between a limited number of recurring structural motifs (e.g., H-bonds, coordination polyhedra, and
protein secondary structure). PAMM (see original publication here) is a clustering technique adapted to the challenges of molecular modeling. This project involves translating the existing implementation of the method, which is written in FORTRAN into more modern, scikit-learn-styled python modules, as part of the scikit-cosmo package.
Auto-Correlation Analysis of Atomistic Simulations
In atomistic simulations, it is non-trivial to determine when a simulation has reached equilibrium — for this one typically employs an order parameter, a behavior or quantity that can be measured and whose convergence signifies the simulation has reached steady-state. In statistical simulations, such order parameters will fluctuate, and it is necessary to de-correlate the results to determine the global statistics. Autocorrelation can also be employed to monitor time-dependent characteristics of the system, such as the mean square displacement as is relevant for diffusion coefficients or dynamic quantities such as the lifetime of a hydrogen-bond. We have developed the toolbox package (https://github.com/cosmo-epfl/toolbox/), a set of C++ routines for efficient computation of autocorrelation analysis. We plan to expand this package in scope and improve its universality. The end goal is the creation of a modular package that can easily interface with popular simulation packages. The ideal student is familiar with C++ and Python, git version control, and understands the basics of molecular dynamics simulations.
Visualizing structure-property relations in materials
In our everyday life, we rely on a relatively small number of materials, with very specific characteristics: ceramics, metallic alloys, polymers, etc.
However, the number of possible materials created by combining different chemical elements in various proportions and arrangements is gigantic.
Because of this, exploring the universe of possible materials to find the best one for a given application is a daunting task, which can be helped by using dimensionality reduction techniques and visualization algorithms.
The student will explore multiple large dataset of materials using different representations and projections to understand how different materials relate to one another; and how we can find good candidates for new applications, either among existing materials or as new, undiscovered materials.
Advanced atomistic simulations: theory and implementation
Materials simulations at the atomic scale rely on advanced statistical mechanics and quantum modelling techniques. Straightforward availability of these approaches in open-source software is highly beneficial for the advancement of our understanding of matter at the atomic scale.
This project – which is particularly suitable for students with little background in atomistic modelling and programming – consists in the study, benchmarking and documentation of one of the methods implemented in the i-PI universal force engine. It should provide with a brief experience in materials modelling, and will teach you how state-of-the-art scientific software is developed and maintained.
A universal engine for the calculation of structural properties of materials
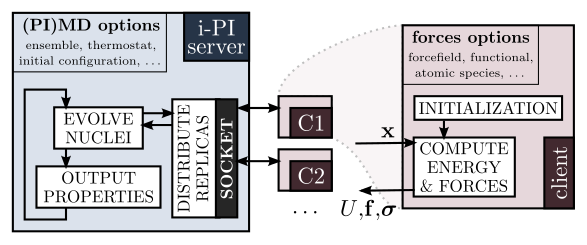
In this project we will extend the functionality of i-PI, a Python interface initially designed for advanced quantum simulations, to perform both routine and advanced calculations of materials’ structural properties (e.g. equilibrium geometry, elastic constants, etc.). i-PI can be easily interfaced to any program designed to compute energy and forces based on empirical potentials or ab initio electronic structure methods, and so these algorithms will be automatically made available to a broad user basis. This project requires some familiarity with atomistic modelling and decent knowledge of programming and Python.
Past Projects
The following items summarize projects that were assigned in the past years. They are not planned as available for the coming semester, but we may discuss possible ways of extending them in case the are of particular interest to you.
Data-driven materials modelling: a primer
More and more often, when modelling the structure-property behavior or materials, expensive electronic-structure calculations are substituted or complemented by a statistical analysis of existing experimental or theoretical data. This project involves the study of some statistical screening techniques, together with the preparation and analysis of appropriate molecular and materials data. Possible systems will be discussed at the start of the project, and include alloys, molecules, and hydrogen-bonded materials.
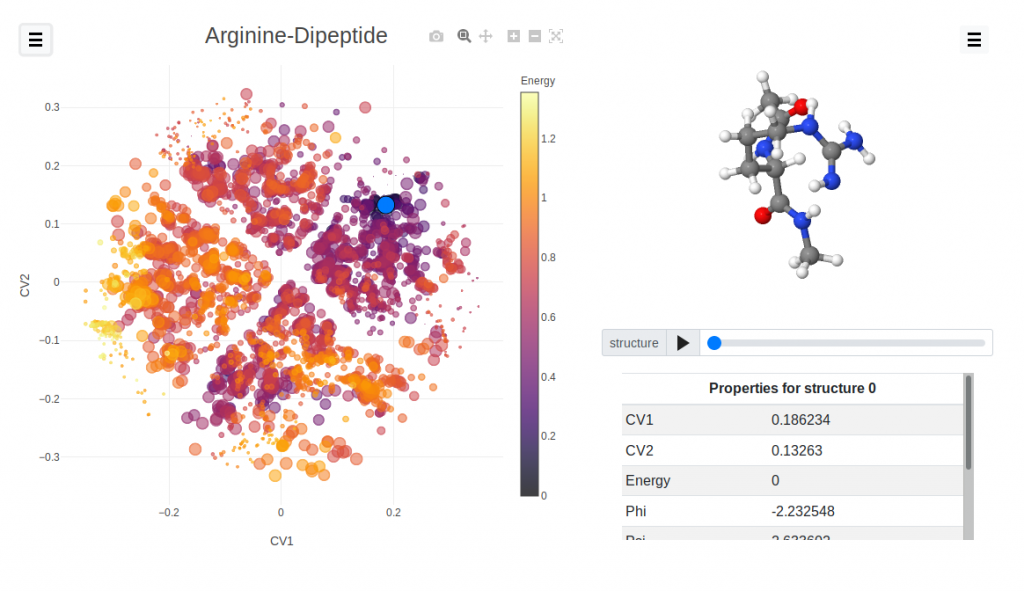
Mapping the stability of oligopeptides
In recent years, a lot of effort has been going on in building atomic/molecular databases containing hundreds and thousands of structures . The aim is to avoid unnecessary replication of work and using machine learning tools to accelerate the discovery of novel materials. With the use of SOAP descriptors combined with Sketchmap algorithm, in our group we have already developed a state of the art technique to visualize and analyze, such large databases. In this project we will analyze such a large public database of oligopeptides ( http://aminoaciddb.rz-berlin. mpg.de/ ) by applying the recently developed methodology, producing a prototype for a web interface to navigate large structural databases.
Atomic pattern matching algorithms in a code for enhanced simulations
The goal of this project is to implement a recently developed strategy to identify recurring atomic patterns in a molecular simulations into PLUMED , a general-purpose plugin to enable advanced molecular simulations in a number of different atomistic codes for the simulations of materials and biomolecules (e.g. LAMMPS, NAMD, GROMACS).
Knowledge of C++ is required, and a degree of familiarity with a LINUX environment (or the MacOS command line) is preferable.
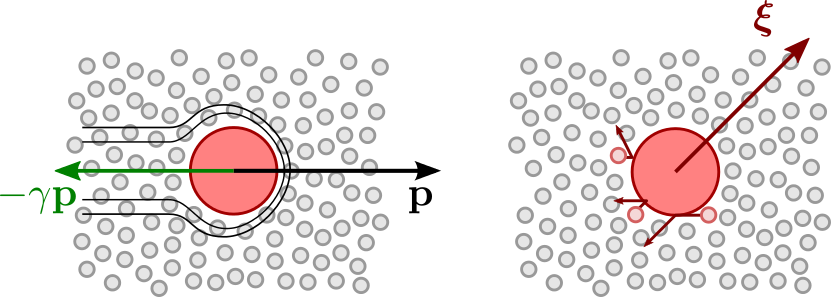
Stochastic methods for atomistic dynamics
Introducing random variables on top of Newtonian dynamics has been used extensively as a method to model materials at constant-temperature thermodynamic conditions. In this project you will learn about the most important of these techniques (that are also used to model the stock market, and other systems characterized by a degree of unpredictable behavior) and modify them to obtain more efficient and physically significant sampling of molecular configurations.
Characterizing the structural complexity of biopolymers using PAMM
The high degree of sophistication found in natural materials is outstanding from the point of view of material science. In a biomaterial the various components of a structure are assembled following a clearly defined pattern: understanding and characterizing such complex patterns is of fundamental importance in order to mimic nature’s ability in designing new materials. In this project we will use a Probabilistic Analysis of Molecular Motifs (PAMM) to analyze recurring patterns in natural peptides, laying the foundations for an analysis of similar structures used as building-blocks of biomimetic polymers.
Characterizing short-range order in atomic systems
Even in highly disordered systems such as liquid or glass, there exists short-range order in the packing of atoms. For instance, neighboring atoms are likely to form icosahedral (fig.), fcc or hcp structures. Although in principle this short-range order can be characterized just by visual inspection, for systems with more than thousands of atoms it is highly desirable to determine it computationally.
The primary goal of the project is to design a simple algorithm to automatically characterize the short-range order of systems, using the input of atomic coordinates.